PIP profiles of coronavirus spike proteins - protein and RNA sequences
Jacques van Helden
2020-05-23
#### General parameters for the analysis ####
## Use (or not) GIDAID sequences
##
## A few genoes were not available in NCBI Genbank at the time of
## this analysis, and had to be downloaded from GISAID. These sequences
## can however not be redistributed, they should thus be downloaded
## manually to reproduce the full trees. Alternatively, useGISAID
## can be set to FALSE, whcih will reproduce the analysis with almost
## all the sequences of the paper.
useGISAID <- TRUE
#### Define directories and files ####
dir <- list(main = '..')
dir$R <- file.path(dir$main, "scripts/R")
#### Create output directory for sequences ####
seqPrefix <- "spike_proteins"
dir$outseq <- file.path(
dir$main, "results", seqPrefix, "PIP_profiles")
dir.create(dir$outseq, showWarnings = FALSE, recursive = TRUE)
# list.files(dir$outseq)
## Instantiate a list for output files
outfiles <- vector()
## Input files
infiles <- list()
#### Sequence collection ####
## Supported:
collections <- c(
"around-CoV-2",
"selected",
"around-CoV-2-plus-GISAID",
"selected-plus-GISAID"
)
## Selected collection
# collection <- "around-CoV-2" # 14 strains
# collection <- "all" # 16 strains
# collection <- "selected" # ~60 strains
# collection <- "around-CoV-2-plus-GISAID" # 16 strains
# collection <- "selected-plus-GISAID" # ~40 strains
collection <- "all-plus-GISAID" # ~60 strains
## Use a collection-specific path for the figures
knitr::opts_chunk$set(
fig.path = paste0('figures/spike-protein_PIP_', collection, '/', collection, "_"))
## Genome dir and files
if (length(grep(pattern = "GISAID", x = collection)) > 0) {
useGISAID <- TRUE
dir$sequences <- file.path(dir$main, "data", "GISAID_genomes")
} else {
dir$sequences <- file.path(dir$main, "data", "genomes")
}
seqPrefix <- paste0("spike_proteins_", collection)
infiles$sequences <- file.path(dir$sequences, paste0(seqPrefix,".fasta"))
## Sequence sequences
if (!file.exists(infiles$sequences)) {
stop("¨Protein sequence file is missing", "\n", infiles$sequences)
}
## Load custom functions
source(file.path(dir$R, "align_n_to_one.R"))
source(file.path(dir$R, "plot_pip_profiles.R"))
## Query patterns for SARS-CoV-2
queryPatterns <- list()
queryPatterns[["HuCoV2_WH01_2019"]] <- c(
"PnGX-P1E_2017",
"PnGu1_2019",
"BtRaTG13_2013_Yunnan",
"BtZC45",
"BtZXC21",
"HuSARS-Frankfurt-1_2003",
"CmMERS",
"HuMERS_172-06_2015"
)
queryPatterns[["PnGu1_2019"]] <- c(
"HuCoV2_WH01_2019",
"PnGX-P1E_2017",
"BtRaTG13_2013_Yunnan"
)
queryPatterns[["BtRaTG13_2013_Yunnan"]] <- c(
"HuCoV2_WH01_2019",
"PnGX-P1E_2017",
"PnGu1_2019"
)
queryPatterns[["HuSARS-Frankfurt-1_2003"]] <- c(
"CvSZ3",
"BtRs4874",
"BtWIV16_2013",
"BtRs4231",
"BtRs7327",
"BtRsSHC014",
"Btrec-SARSp_2008",
"PnGX-P1E_2017",
"HuCoV2_WH01_2019",
"BtRaTG13_2013_Yunnan",
"BtZC45",
"BtZXC21",
"CmMERS",
"HuMERS_172-06_2015"
)
#### Add GISAID IDs to the query pattern ####
## Note that GISAID sequences are be submitted to the github repo because they cannot be redistributed
if (useGISAID) {
for (ref in names(queryPatterns))
queryPatterns[[ref]] <- append(queryPatterns[[ref]],
c("BtYu-RmYN02_2019",
"PnGu1_2019"
))
}
# message("\tReference strain: ", refPattern)
message("\tQuery patterns")
for (ref in names(queryPatterns)) {
queryPatterns[[ref]] <- unique(queryPatterns[[ref]])
message("\t", ref, "\t\t", length(queryPatterns[[ref]]),
"\t", paste(collapse = ", ", queryPatterns[[ref]]))
}#### Load sequences ####
sequences <- readAAStringSet(filepath = infiles$sequences, format = "fasta")
## Shorten sequence names by suppressing the fasta comment (after the space)
names(sequences) <- sub(pattern = " .*", replacement = "", x = names(sequences), perl = TRUE)
sequencesNames <- names(sequences)
nbsequences <- length(sequencesNames)
message("Loaded ", nbsequences, " sequences from file ", infiles$sequences)
# View(sequences)
#### Define reference and query sequences ####
refSequenceNames <- vector()
for (ref in names(queryPatterns)) {
refSequenceNames[ref] <- unique(
grep(pattern = ref, x = names(sequences),
ignore.case = TRUE, value = TRUE))
if (is.null(refSequenceNames[ref])) {
stop("Could not identify reference sequences with pattern ", ref)
}
}
## Query sequences
# querySequenceNames <- list()
# for (ref in names(queryPatterns)) {
# # message("Identifying query sequences for reference: ", ref)
# queryRegExp <- paste0("(", paste(collapse = ")|(", queryPatterns[[ref]]), ")")
# querySequenceNames[[ref]] <- grep(pattern = queryRegExp,
# x = sequencesNames,
# ignore.case = TRUE, value = TRUE)
# nbquerySeq <- length(querySequenceNames[[ref]])
#
# if (nbquerySeq == 0) {
# stop("Could not identify any query sequences with query pattern\n", queryRegExp)
# }
#
# if (length(unlist(queryPatterns[ref[]])) != length(querySequenceNames[[ref]])) {
# foundPatterns <- grep(pattern = queryRegExp, x = querySequenceNames[[ref]], value = TRUE)
# missingPatterns <- setdiff(foundPatterns, queryPatterns[[ref]])
# message("\t",
# length(missingPatterns), " Missing sequences: ",
# paste(collapse = ", ", missingPatterns))
# }
#
# message("\tFound ",
# "\t", length(querySequenceNames[[ref]]),
# " queries for\t", ref)
#
# ## Compute some statistics about sequences sizes
# sequencestat <- data.frame(
# row.names = c(ref, querySequenceNames[[ref]]),
# status = c("Reference",
# rep("Query", length.out = length(querySequenceNames[[ref]])))
# )
#
# g <- 1
# for (g in c(ref, querySequenceNames[[ref]])) {
# sequencestat[g, "length"] <- length(sequences[[g]])
# }
# kable(sequencestat, caption = paste0("Query sequences for reference ", ref))
# }The collection all-plus-GISAID contains 67 virus sequences sequences.
## Report the number of strains
strainNames <- names(sequences)
nbStrains <- length(strainNames)
message("\tLoaded ", nbStrains, " sequences from file ", infiles$sequences)
# View(genomes)
#### Compute sequence sizes ####
strainStats <- data.frame(
n = 1:length(sequences),
row.names = names(sequences),
status = rep("Query", length.out = length(strainNames))
)
strainStats[,"status"] <- as.vector(strainStats[,"status"])
strainStats[refSequenceNames,"status"] <- "Reference"
g <- 1
for (g in strainNames) {
strainStats[g, "length"] <- length(sequences[[g]])
}
#### Define the color associated to each sequence ####
## Color palette per species
speciesPalette <- list(
Human = "#880000",
Bat = "#888888",
Pangolin = "#448800",
Camel = "#BB8800",
Pig = "#FFBBBB",
Civet = "#00BBFF"
)
## Species prefix in the tip labels
speciesPrefix <- c("Hu" = "Human",
"Bt" = "Bat",
"Pn" = "Pangolin",
"Cm" = "Camel",
"Pi" = "Pig",
"Cv" = "Civet")
## Strain-specific colors
strainColor <- c(
"HuCoV2_WH01_2019" = "red",
"HuSARS-Frankfurt-1_2003" = "#0044BB",
"BtRs4874" = "#BB00BB",
"PnGu1_2019" = "#00BB00",
"BtRaTG13_" = "#FF6600",
"BtYu-RmYN" = "#FFBB22",
"BtZXC21" = "black",
"BtZC45" = "black")
## Identify species per tip
for (prefix in names(speciesPrefix)) {
strainStats[grep(pattern = paste0("^", prefix), x = row.names(strainStats), perl = TRUE), "species"] <- speciesPrefix[prefix]
}
## Assign acolor to each species
strainStats$color <- "grey" # default
strainStats$color <- speciesPalette[as.vector(strainStats$species)]
for (strain in names(strainColor)) {
strainStats[grep(pattern = paste0("^", strain),
x = row.names(strainStats), perl = TRUE), "color"] <- strainColor[strain]
}
## Assign specific color to some nodes
## Define a color for each strain
strainColors <- (unlist(strainStats$color))
names(strainColors) <- row.names(strainStats)N-to-1 full sequence alignments
We perform a pairwise lignment between each sequences query and the reference sequences (HuCoV2_WH01_2019).
sequencesNto1 <- list()
refSequenceName <- "HuCoV2_WH01_2019"
for (refSequenceName in refSequenceNames) {
## Define output file for sequences alignments
outfile <- file.path(
dir$outseq, paste0("one-to-n_alignments_ref_", refSequenceName))
outfiles[paste0(refSequenceName, " alignments")] <- outfile
## Get sequences for reference and query sequences
refSequence <- sequences[refSequenceName]
# querySeq <- sequences[querySequenceNames[[refSequenceName]]]
message("\tAligning ", length(sequences), " sequences",
" to reference\t", refSequenceName)
sequencesNto1[[refSequenceName]] <- alignNtoOne(
refSequence = refSequence,
querySequences = sequences,
# querySequences = querySeq,
sortByPIP = TRUE,
# querySequences = sequences,
outfile = outfile)
}refSequenceName <- "HuCoV2_WH01_2019"
for (refSequenceName in refSequenceNames) {
cat(sep = "", "\n## Spike proteins: ", collection, " vs reference ", refSequenceName)
knitr::opts_chunk$set(
fig.path = paste0('figures/spike-protein_PIP/', collection, '_vs_', refSequenceName))
kable(sequencesNto1[[refSequenceName]]$stats[order(sequencesNto1[[refSequenceName]]$stats$score, decreasing = TRUE), ],
caption = "N-to-one alignment of full sequences")
## PIP profile of full sequences N-to-1 alignments
if (length(grep(pattern = "around-CoV-2", x = collection))) {
minPIP <- 30
legendCex <- 1
legendCorner <- "bottomright"
legendMargin <- 0
} else {
minPIP <- 0
legendCex <- 0.65
legendCorner <- "topright"
legendMargin <- 0.25
}
plotPIPprofiles(
alignments = sequencesNto1[[refSequenceName]]$alignments,
windowSize = 100,
main = paste0("Spike protein: collection ", collection, "\nvs strain ", refSequenceName),
colors = strainColors,
legendMargin = legendMargin,
legendCorner = legendCorner,
lwd = 1,
legendCex = legendCex,
ylim = c(minPIP,100))
# legendMargin = 0.25,
# legendCorner = "topright", lwd = 1,
# legendCex = 0.5, ylim = c(30, 100))
}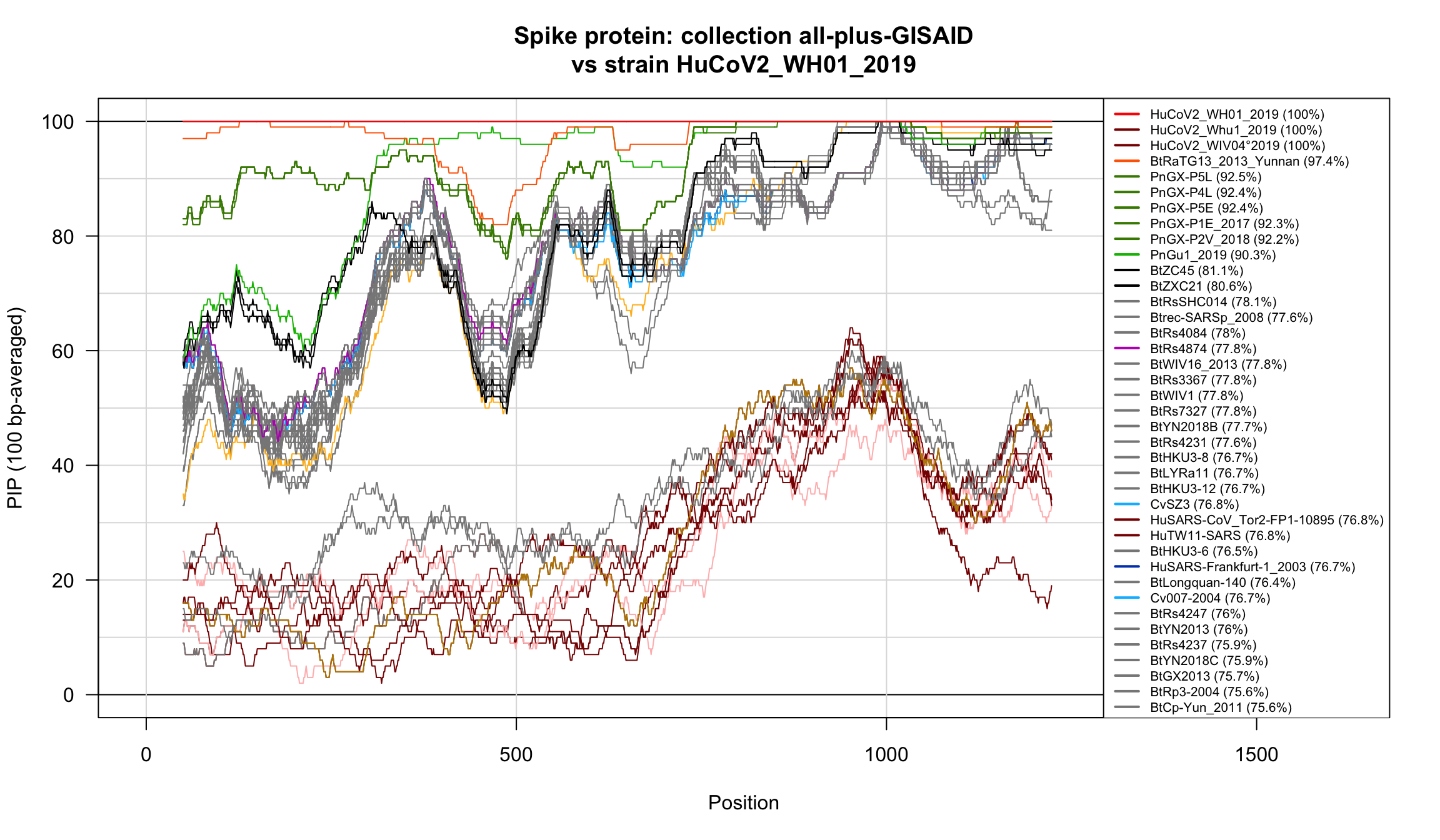
Percent Identical Positions (PIP) profiles of spike protein sequences.
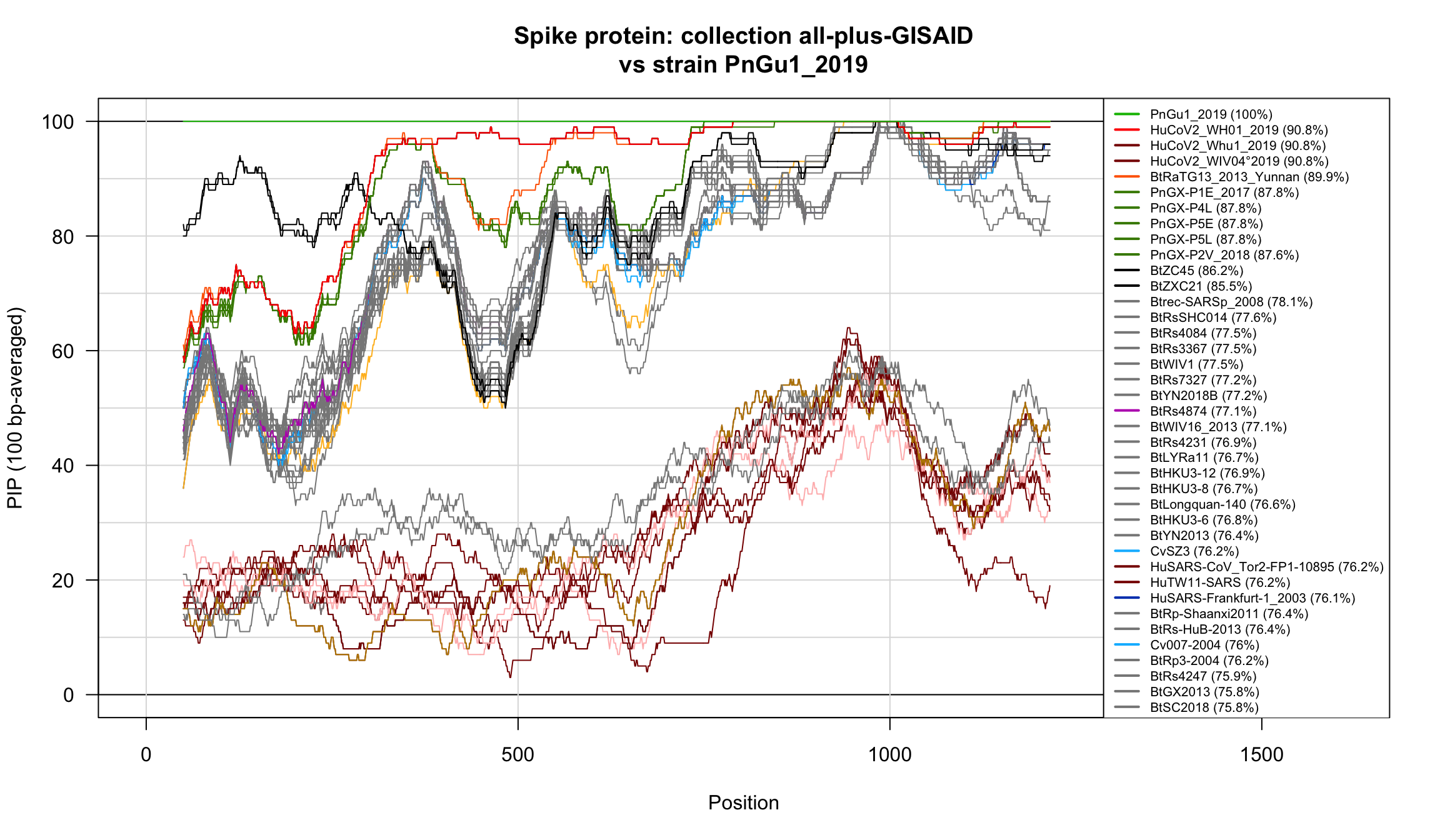
Percent Identical Positions (PIP) profiles of spike protein sequences.
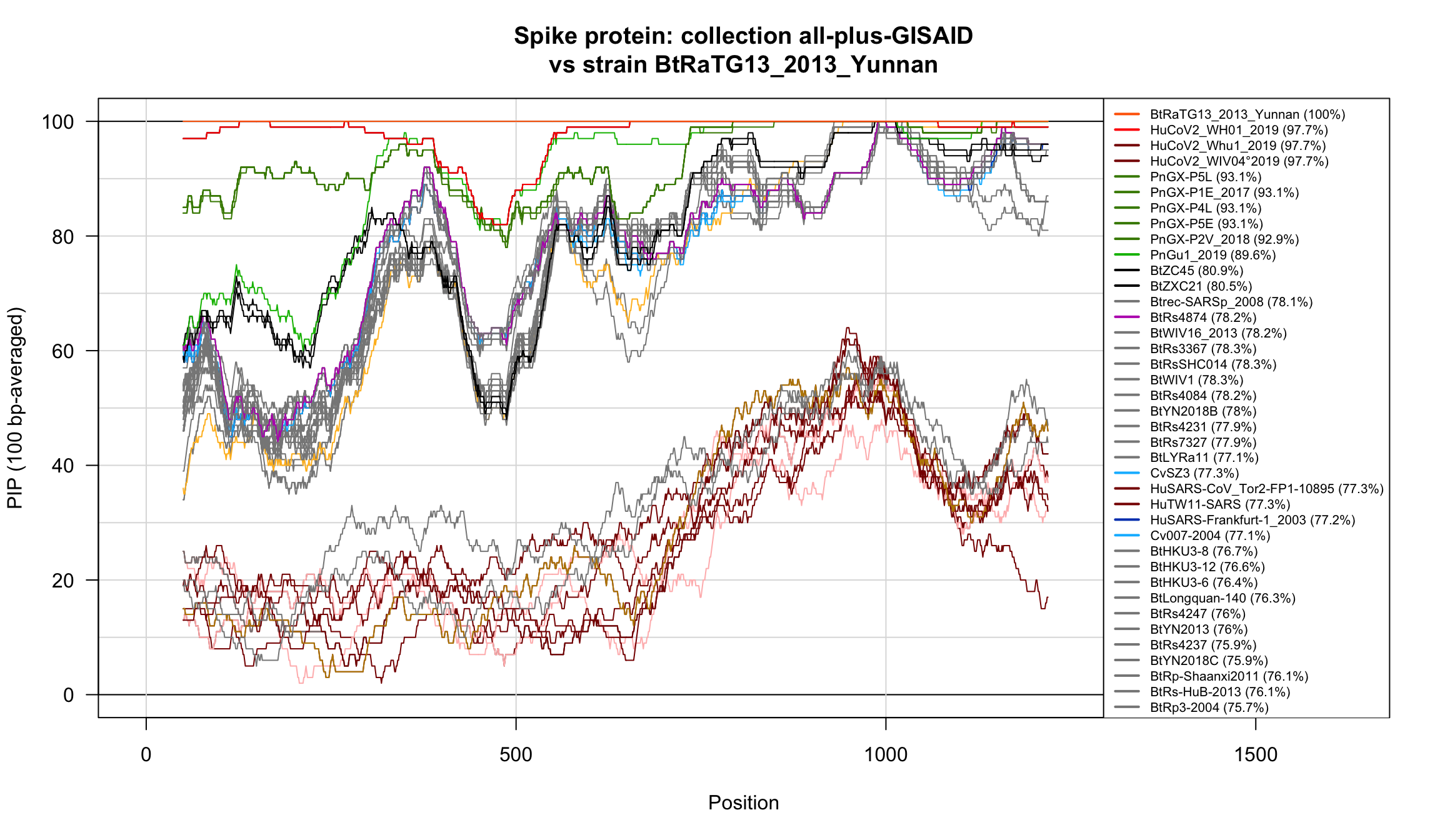
Percent Identical Positions (PIP) profiles of spike protein sequences.
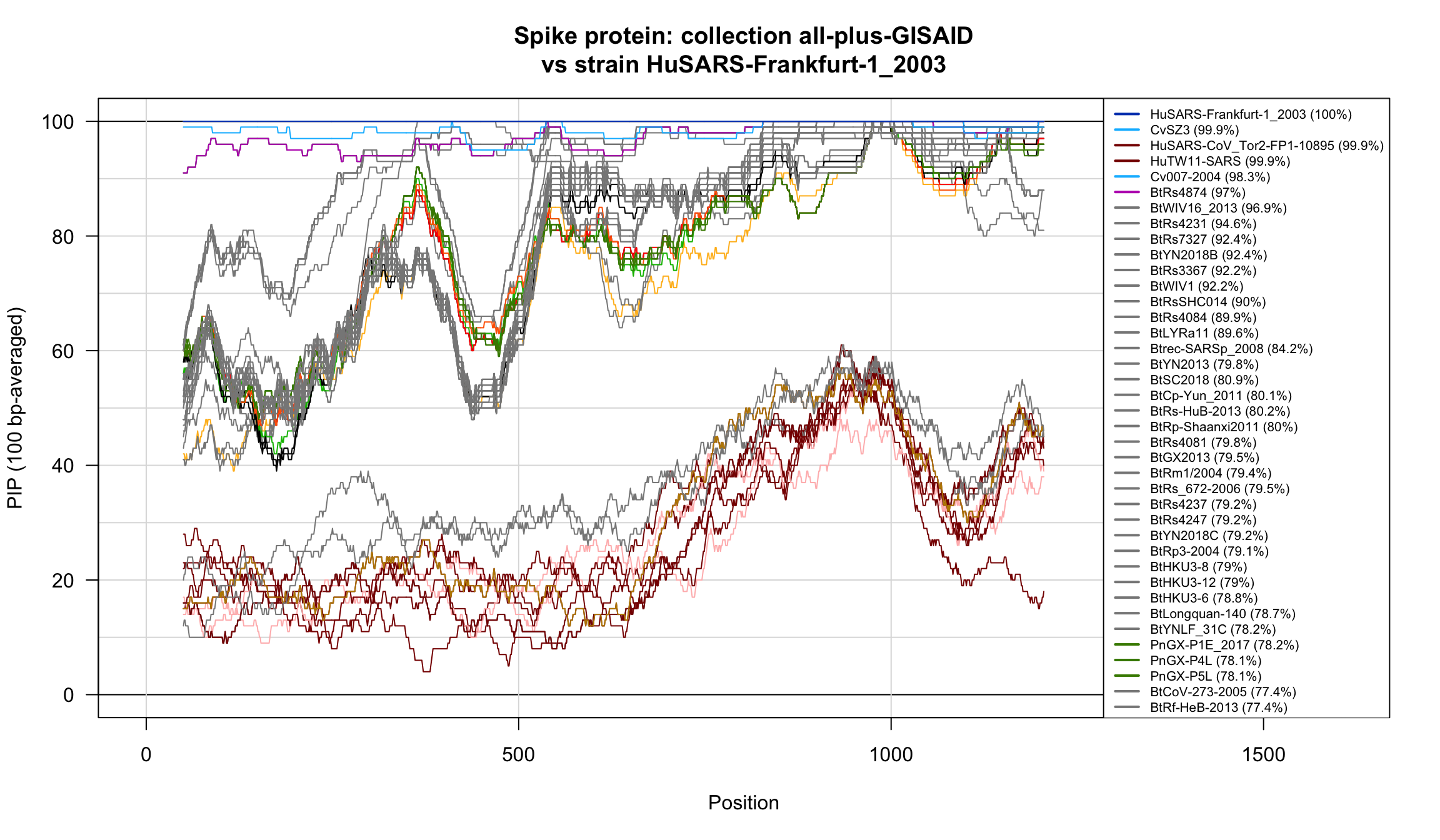
Percent Identical Positions (PIP) profiles of spike protein sequences.
Output files
| Dir | |
|---|---|
| main | .. |
| R | ../scripts/R |
| outseq | ../results/spike_proteins/PIP_profiles |
| sequences | ../data/GISAID_genomes |
| File | |
|---|---|
| HuCoV2_WH01_2019 alignments | ../results/spike_proteins/PIP_profiles/one-to-n_alignments_ref_HuCoV2_WH01_2019 |
| PnGu1_2019 alignments | ../results/spike_proteins/PIP_profiles/one-to-n_alignments_ref_PnGu1_2019 |
| BtRaTG13_2013_Yunnan alignments | ../results/spike_proteins/PIP_profiles/one-to-n_alignments_ref_BtRaTG13_2013_Yunnan |
| HuSARS-Frankfurt-1_2003 alignments | ../results/spike_proteins/PIP_profiles/one-to-n_alignments_ref_HuSARS-Frankfurt-1_2003 |
Session info
R version 3.6.1 (2019-07-05)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] Biostrings_2.52.0 XVector_0.24.0 IRanges_2.18.3 S4Vectors_0.22.1 BiocGenerics_0.30.0 knitr_1.28
loaded via a namespace (and not attached):
[1] Rcpp_1.0.4 digest_0.6.25 magrittr_1.5 evaluate_0.14 highr_0.8 zlibbioc_1.30.0 rlang_0.4.5 stringi_1.4.6 rmarkdown_2.1 tools_3.6.1 stringr_1.4.0 xfun_0.12 yaml_2.2.1 compiler_3.6.1
[15] BiocManager_1.30.10 htmltools_0.4.0