Phylogenetic analysis of coronavirus sequences
Jacques van Helden
2021-02-15
## Color palette per species
speciesPalette <- list(
Human = "#880000",
Bat = "#888888",
Pangolin = "#448800",
Camel = "#BB8800",
Pig = "#FFBBBB",
Civet = "#00BBFF"
)
## Species prefix in the tip labels
speciesPrefix <- c("Hu" = "Human",
"Bt" = "Bat",
"Pn" = "Pangolin",
"Cm" = "Camel",
"Pi" = "Pig",
"Cv" = "Civet")
## Strain-specific colors
strainColor <- c(
"HuCoV2" = "red",
"HuSARS-Fr" = "#0044BB",
"PnGu1" = "#00BB00",
"BtRaTG13" = "#FF6600",
"BtYu-RmYN" = "#FFBB22",
"BtZXC21" = "black",
"BtZC45" = "black")
## Define feature types
features <- c(
# "genomes",
# "S-gene",
"S1",
"S2",
"RBD",
# "Recomb-Xiao",
"Recomb-reg-1",
"Recomb-reg-2",
"Recomb-reg-3",
"CDS-ORF1ab",
"After-ORF1ab",
"CDS-S",
"CDS-ORF3a",
"CDS-E",
"CDS-M",
"CDS-ORF6",
"CDS-ORF7a",
"CDS-ORF8",
"CDS-N",
"CDS-ORF10")
feature <- "S-gene"
## Define collections
collections <- c("around-CoV-2", "selected")
collection <- "around-CoV-2" # default for testing
## Outgroup per collection
outgroups <- list()
outgroups[["selected"]] <- c(
"HuOC43",
"PiPRCV",
"HuTGEV",
"PiSADS",
"Hu229E",
"HuNL63")
outgroups[["around-CoV-2"]] <- "BtBM48-31"
## Use GISAID data
useGISAID <- TRUE
if (useGISAID) {
collections <- paste0(collections, "-plus-GISAID")
for (collection in names(outgroups)) {
outgroups[[paste0(collection, "-plus-GISAID")]] <- outgroups[[collection]]
}
collection <- paste0(collection, "-plus-GISAID")
}dir <- vector()
dir["main"] <- ".."
dir["results"] <- file.path(dir["main"], "results")
dir["genomes"] <- file.path(dir["results"], "genome_phylogeny", "clustalw_alignments")
dir["R"] <- file.path(dir["main"], "scripts", "R")
# list.files(dir["R"])
source(file.path(dir["R"], "load_tree.R"))
source(file.path(dir["R"], "plot_my_tree.R"))Phylogeny from full genomes
We inferred a phylogeny of virus strains based ontheir full genomes.
A multiple alignment of genome sequences was performed with a progressive method (clustalw). The tree of virus strains was inferred with a maximum likelihood approach (phyml software).
#### Load and plot the genome tree ####
genomeTreeFile <- file.path(
dir["genomes"],
"coronavirus_selected-plus-GISAID_genomes_clustalw_gblocks.phy_phyml_tree.phb")
genomeTree <- loadTree(
treeFile = genomeTreeFile,
outgroup = outgroups[['selected']],
rootNode = NULL,
speciesPalette = speciesPalette,
tipColor = strainColor,
nodesToRotate = c(39, 75, 42))
# genomeTree <- paintSubTree(tree = genomeTree, node = 49, state = "CoV2")
plotMyTree(genomeTree, main = "Genome-based virus tree",
scaleLength = 0.1,
show.node.label = FALSE)
# nodelabels(cex = 0.4)
## Identify some clades
cladelabels(genomeTree$tree, "CoV2", 46, cex = 0.7, orientation = "horizontal", offset = 5)
cladelabels(genomeTree$tree, "MERS", 43, cex = 0.7, orientation = "horizontal", offset = 5)
cladelabels(genomeTree$tree, "SARS", 69, cex = 0.7, orientation = "horizontal", offset = 5)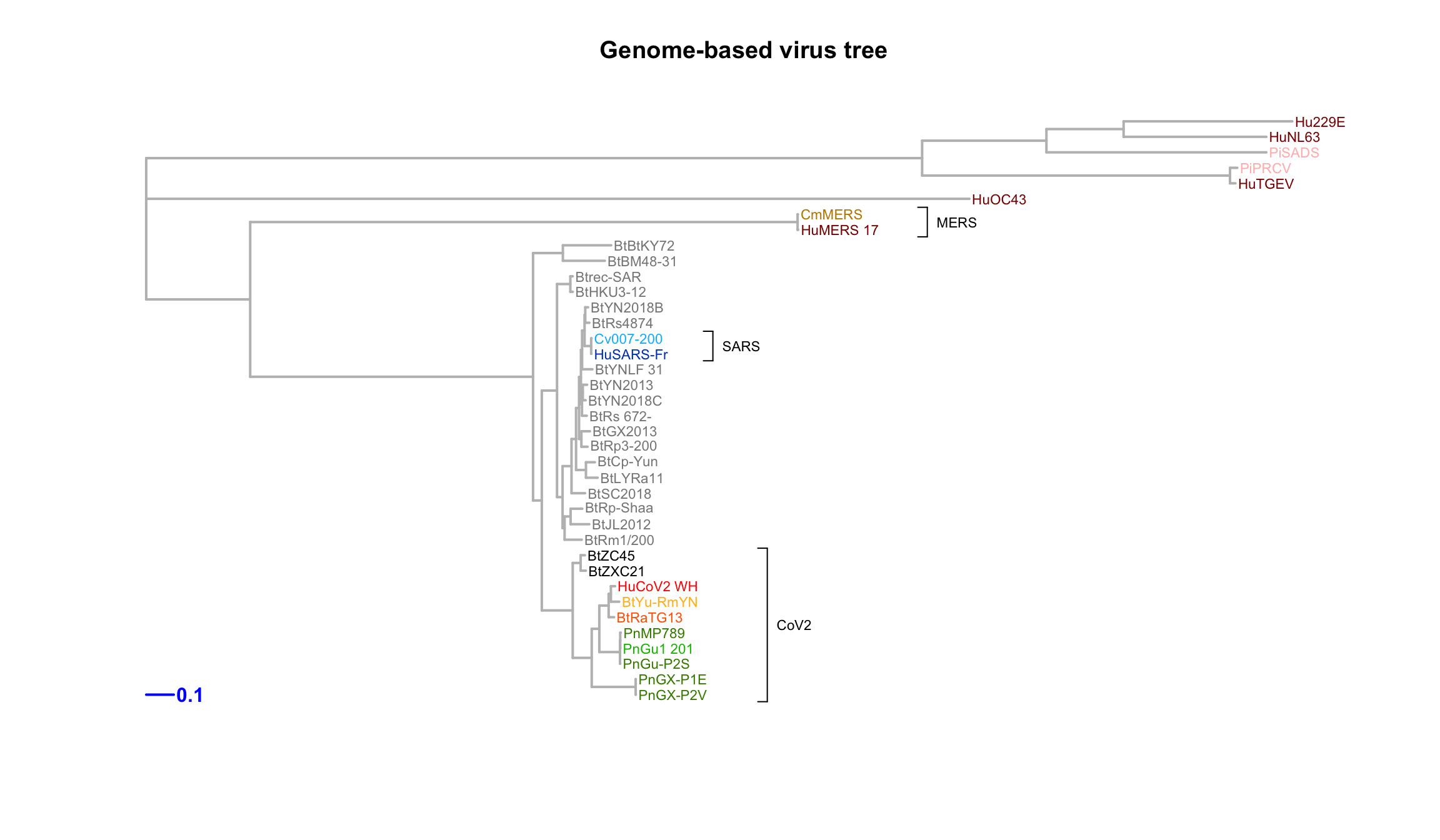
Genome tree of selected coronaviruses. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
Tree per feature
#### Load and plot feature-specific trees ####
## Define vectors to hold the results and enable tree comparisons
treeFiles <- vector()
treeData <- list()
## Define the path to the tre file
feature <- "RBD"
for (feature in features) {
cat(" \n### ", feature, "\n")
message("\n\tReading tree for feature ", feature)
prefix <- paste0(feature, "_", collection)
treeFile <- file.path(
dir["results"],
prefix,
paste0(prefix, "_clustalw_gblocks.phy_phyml_tree_GTR.phb"))
treeFiles[feature] <- treeFile
## Load the tree
treeData[[feature]] <- loadTree(
treeFile = treeFiles[feature],
outgroup = outgroups[[collection]],
rootNode = NULL,
speciesPalette = speciesPalette,
tipColor = strainColor,
nodesToRotate = NULL)
plotMyTree(treeData[[feature]],
main = paste0(feature, " tree"),
scaleLength = 0.05, cex = 1, label.offset = 0.01,
show.node.label = FALSE)
# ## Identify some clades
# cladelabels(genomeTree$tree, "CoV2", 46, cex = 0.7, orientation = "horizontal", offset = 5)
# cladelabels(genomeTree$tree, "MERS", 43, cex = 0.7, orientation = "horizontal", offset = 5)
# cladelabels(genomeTree$tree, "SARS", 69, cex = 0.7, orientation = "horizontal", offset = 5)
}S1
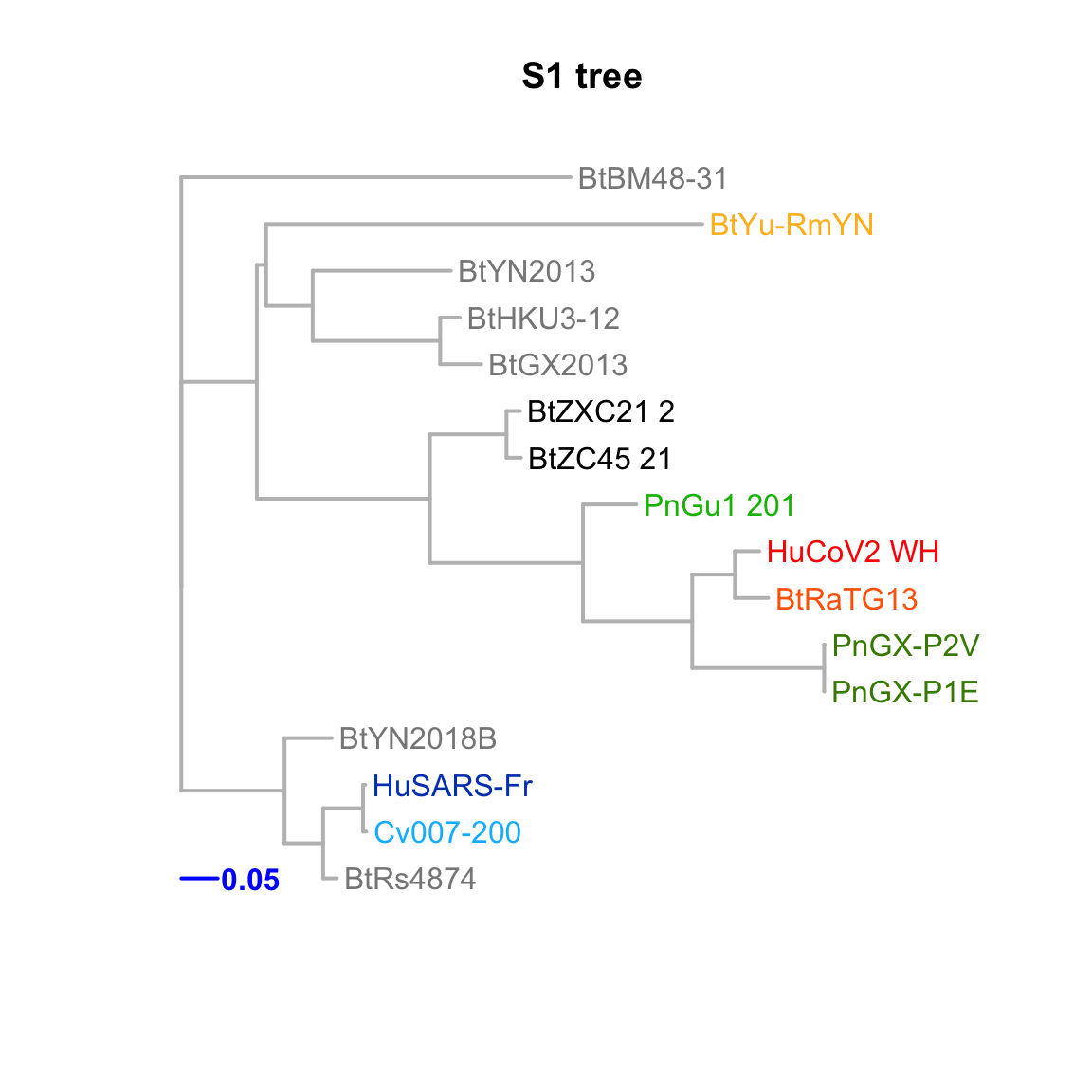
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
S2
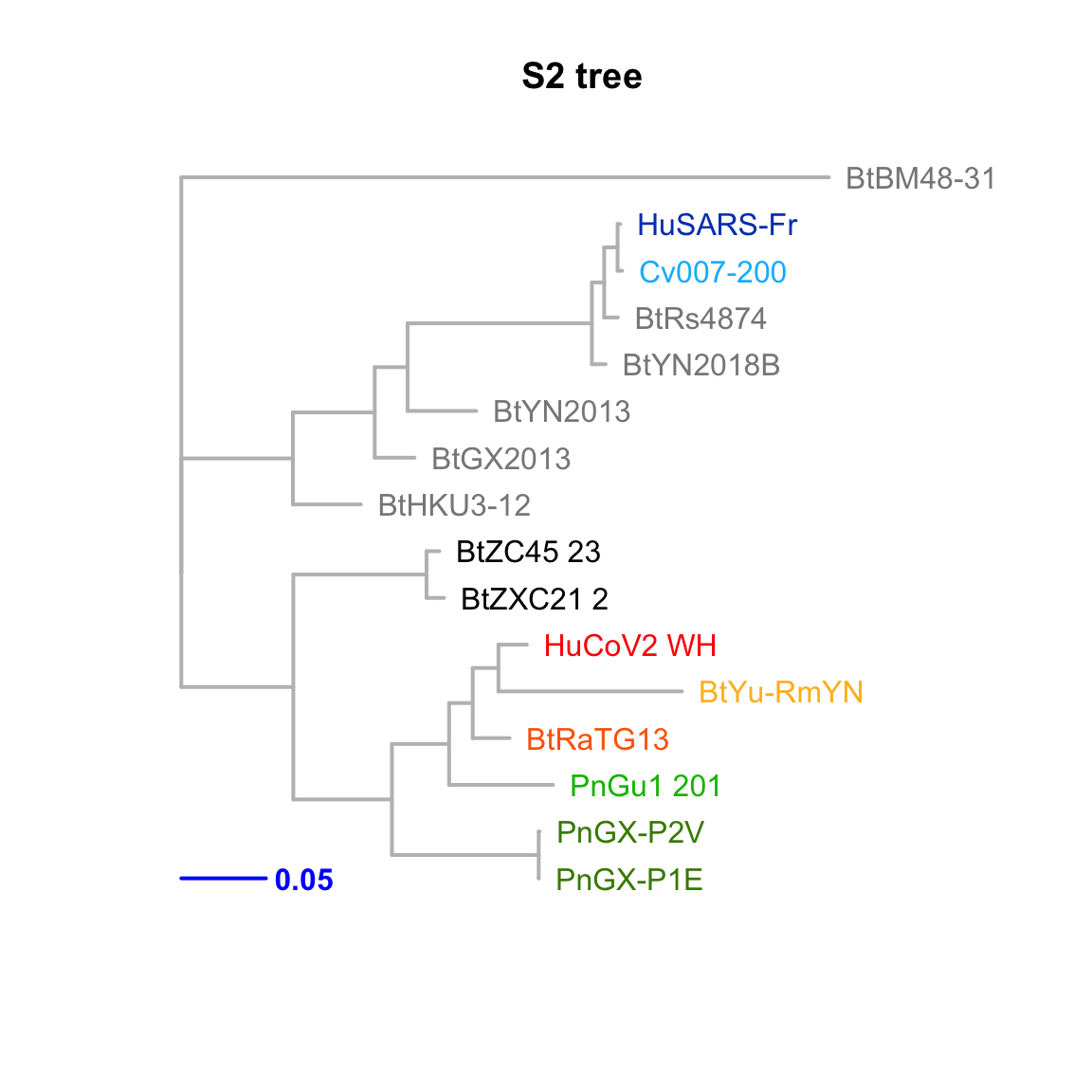
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
RBD
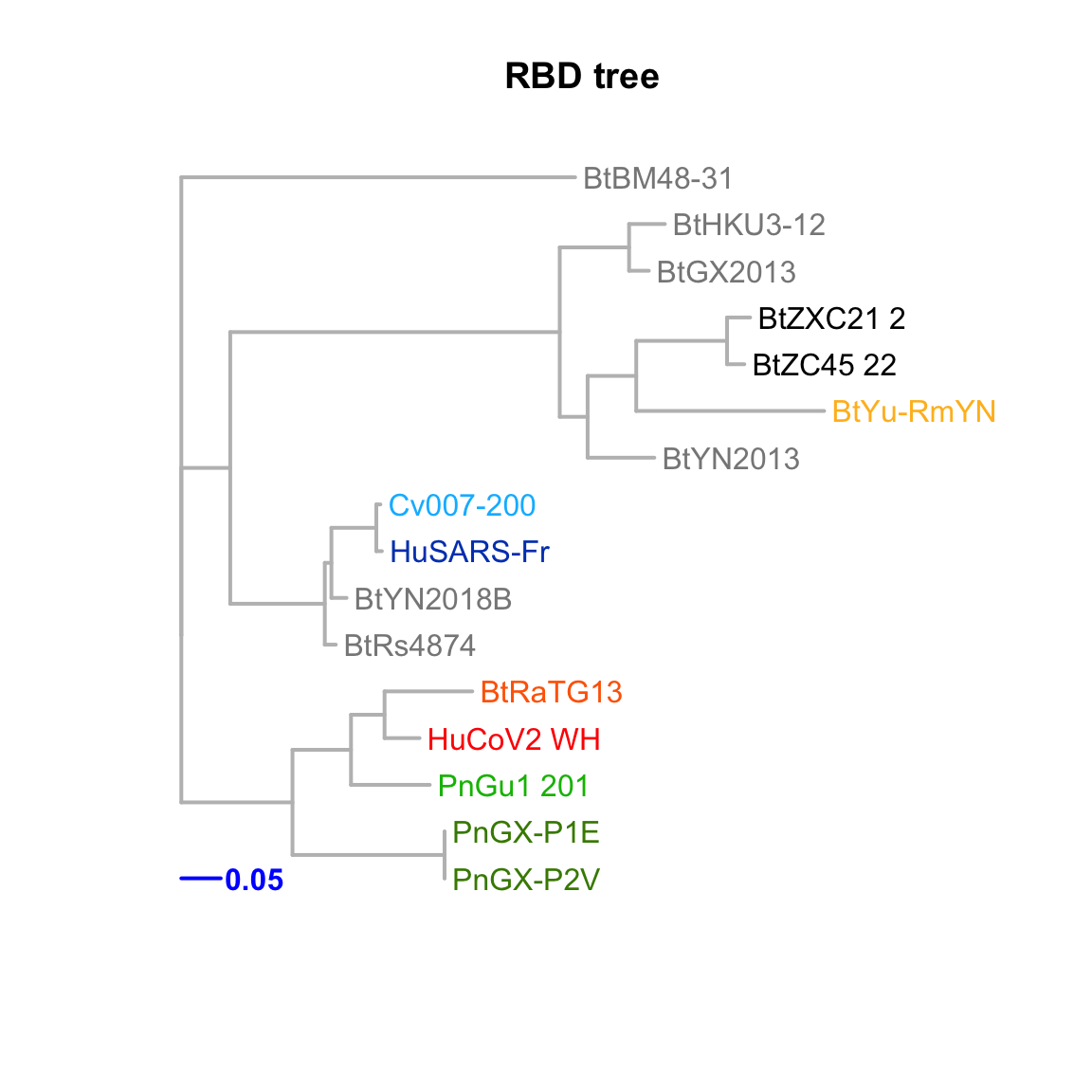
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
Recomb-reg-1
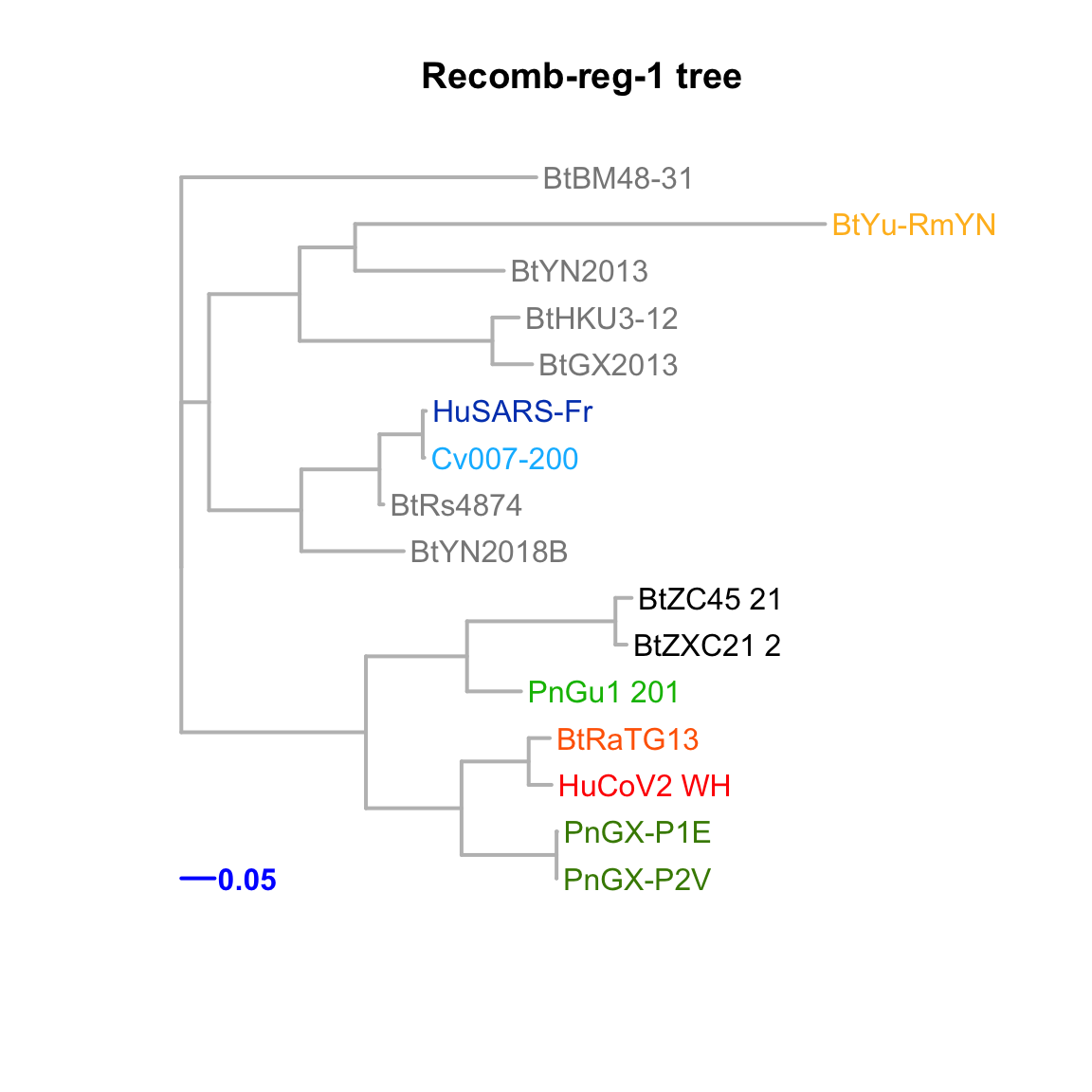
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
Recomb-reg-2
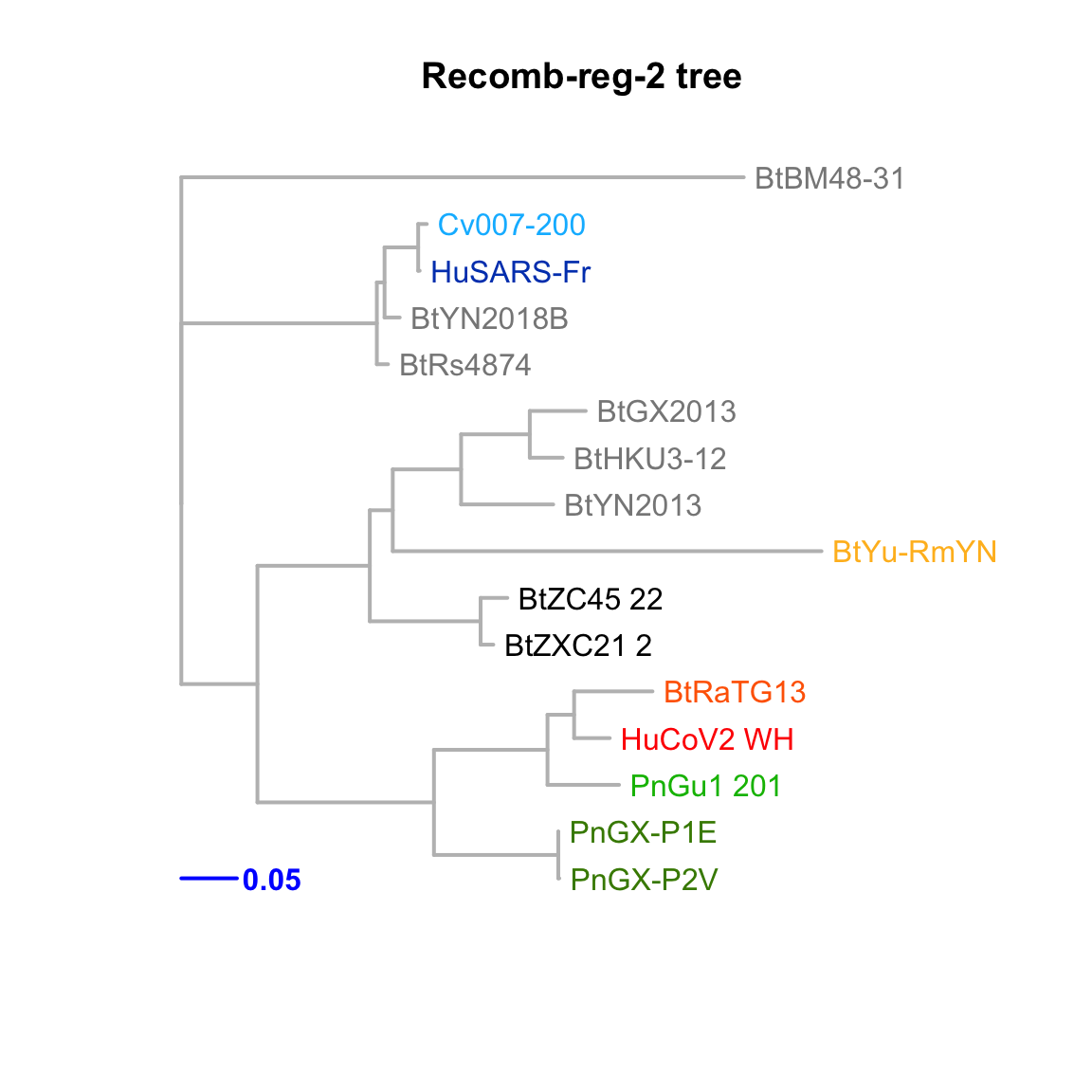
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
Recomb-reg-3
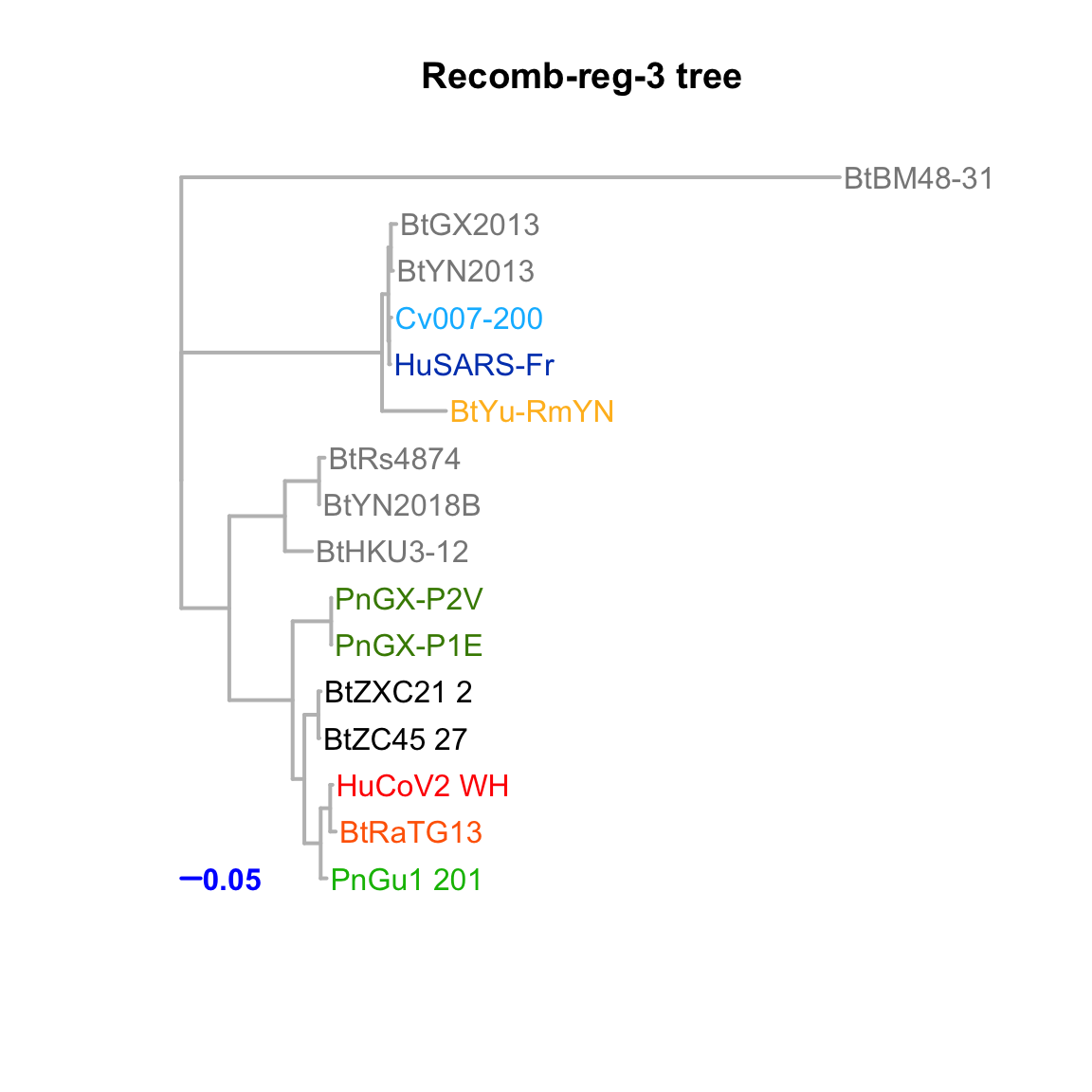
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF1ab
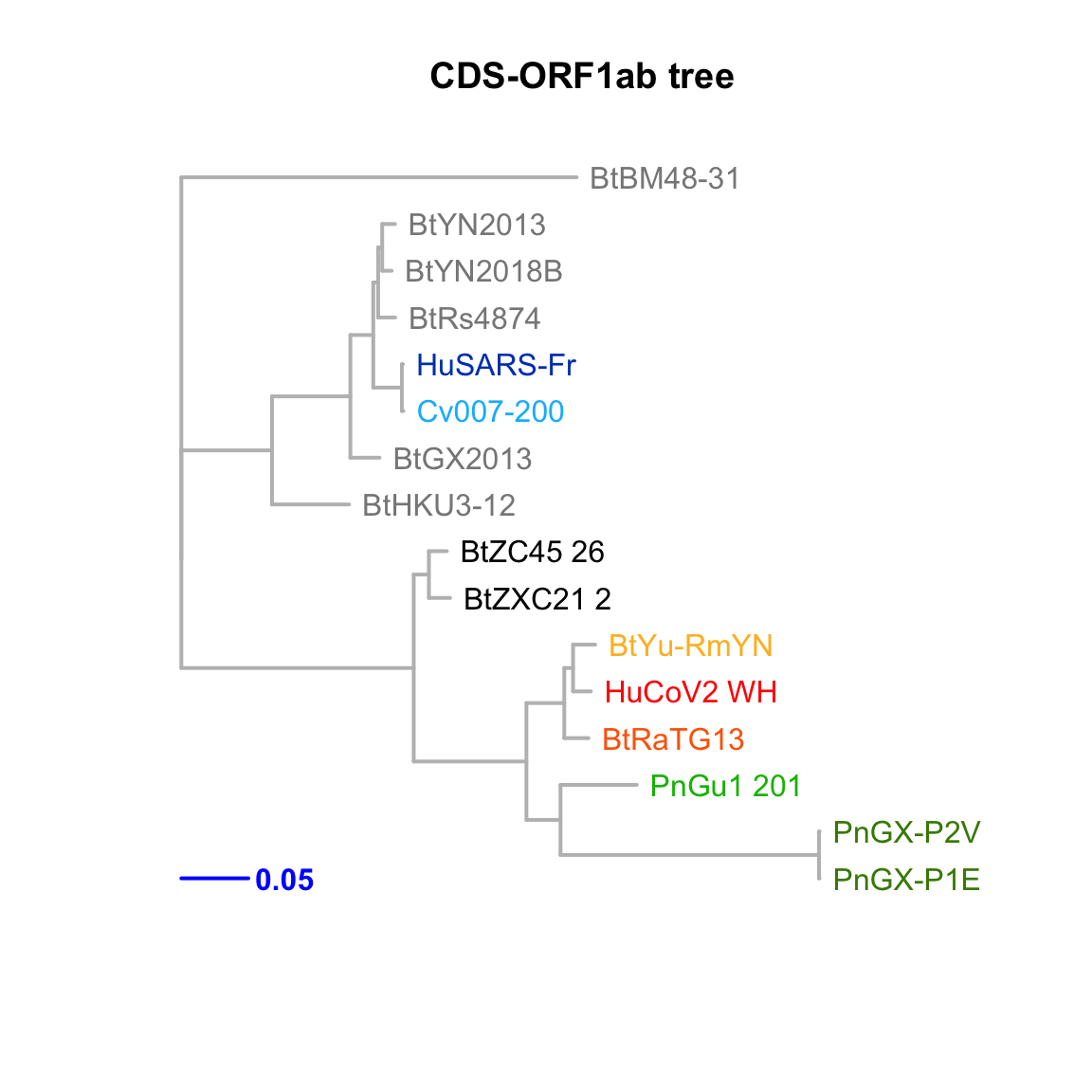
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
After-ORF1ab
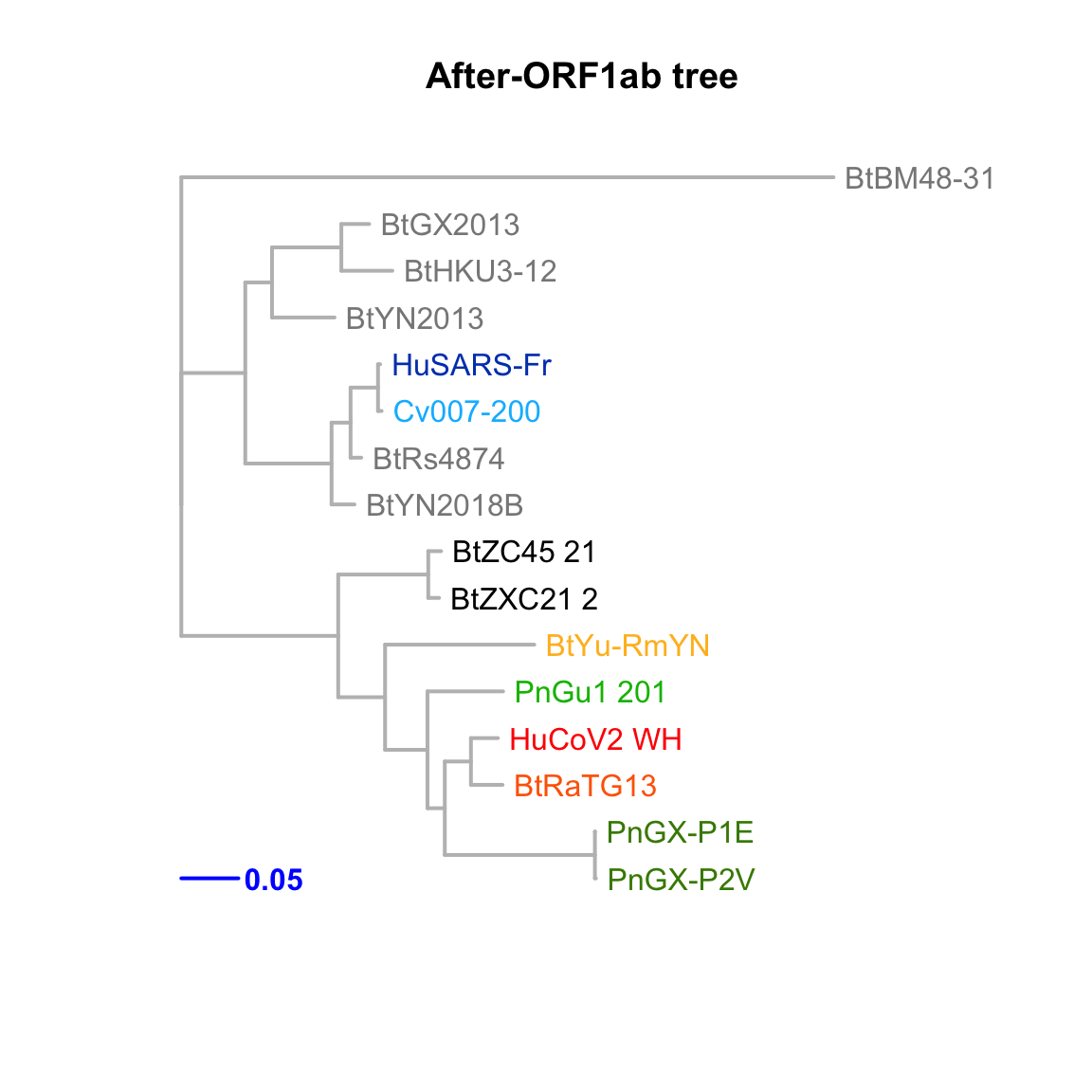
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-S
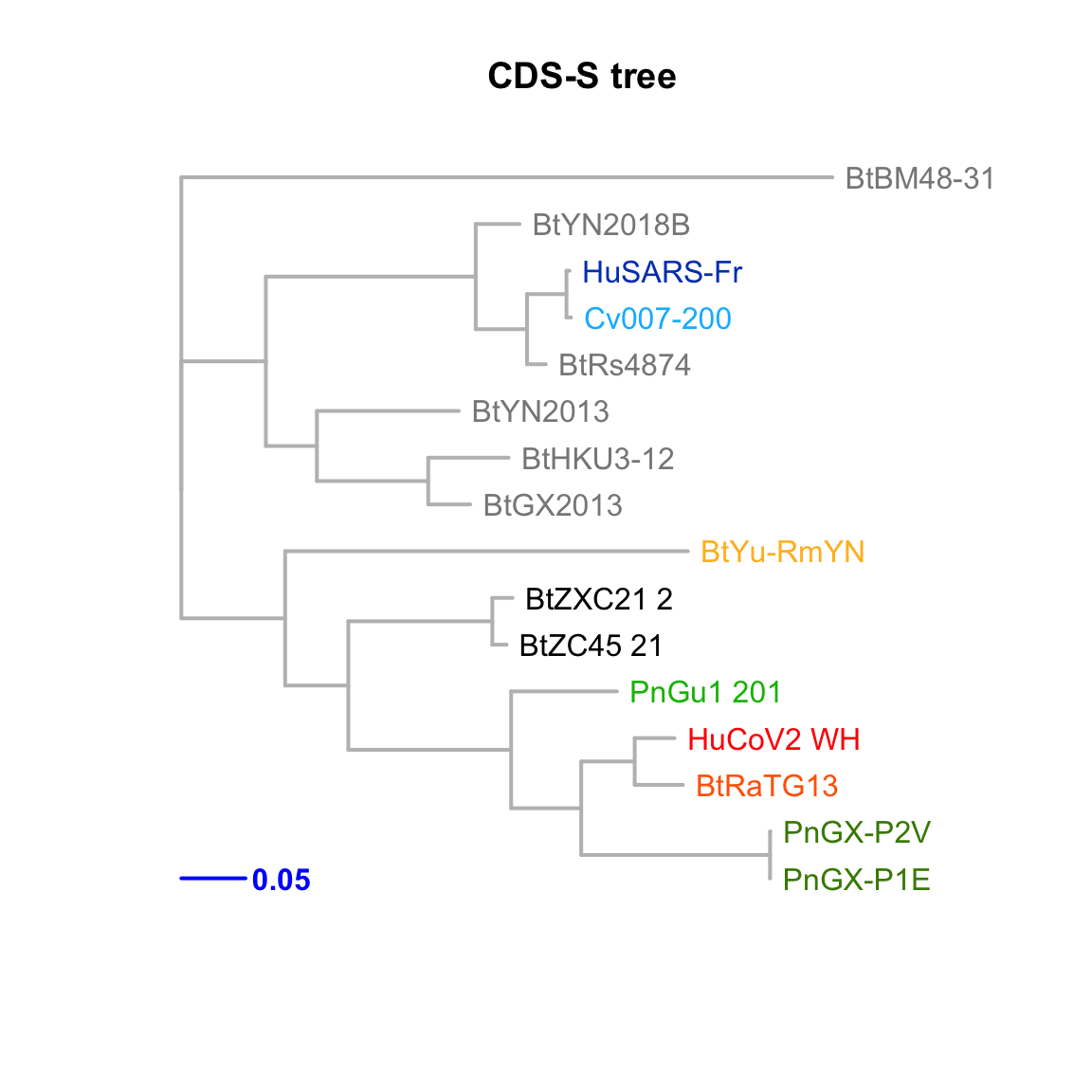
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF3a
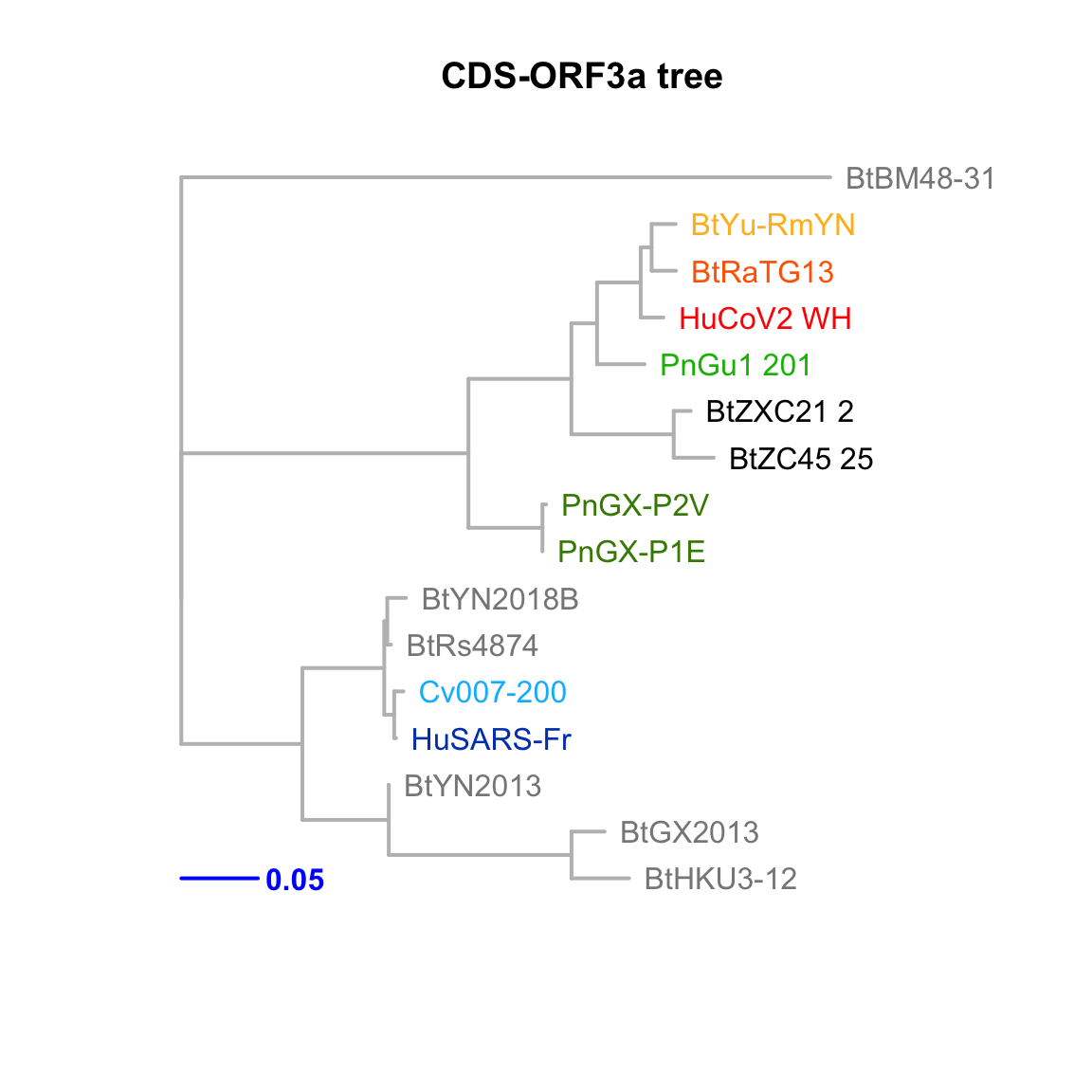
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-E
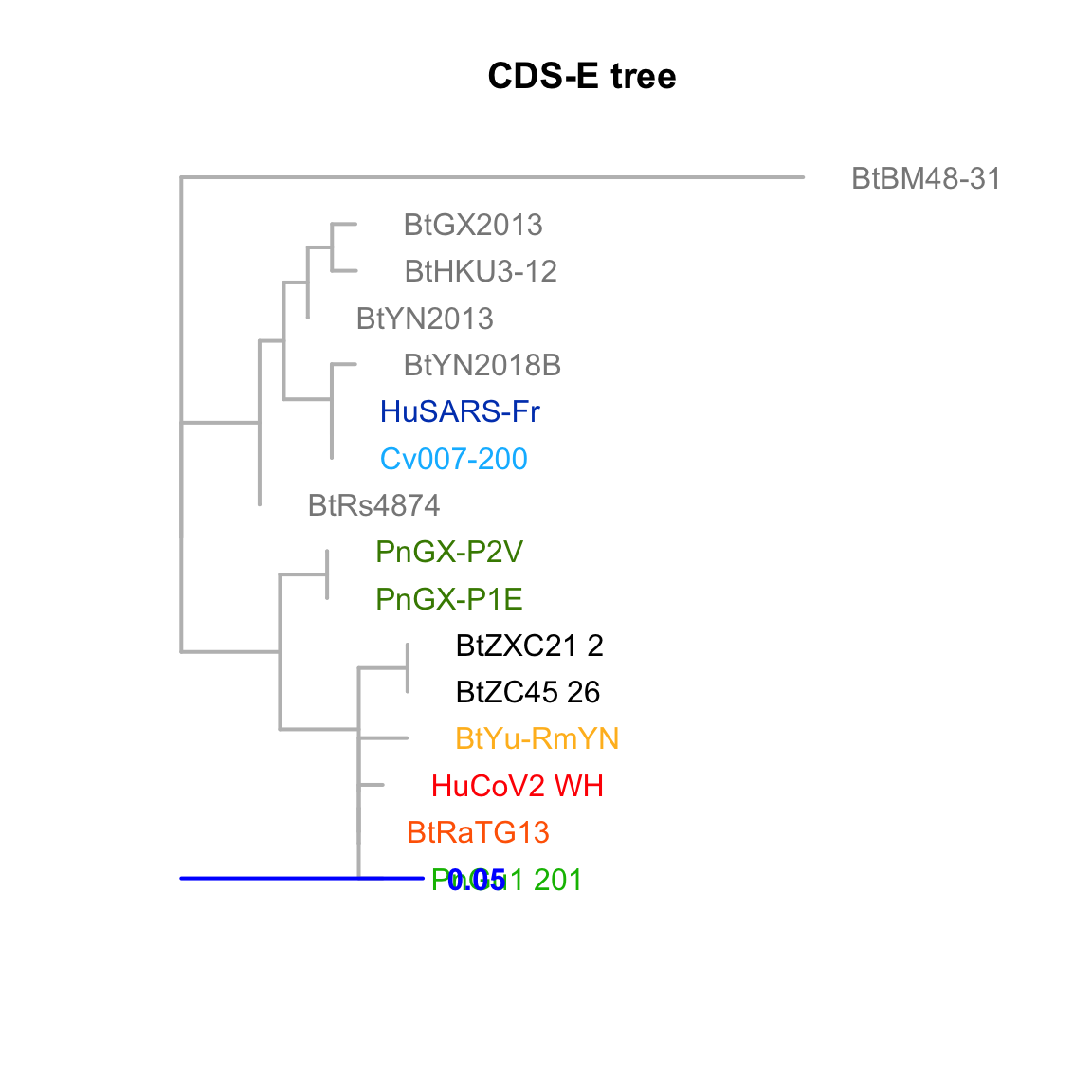
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-M
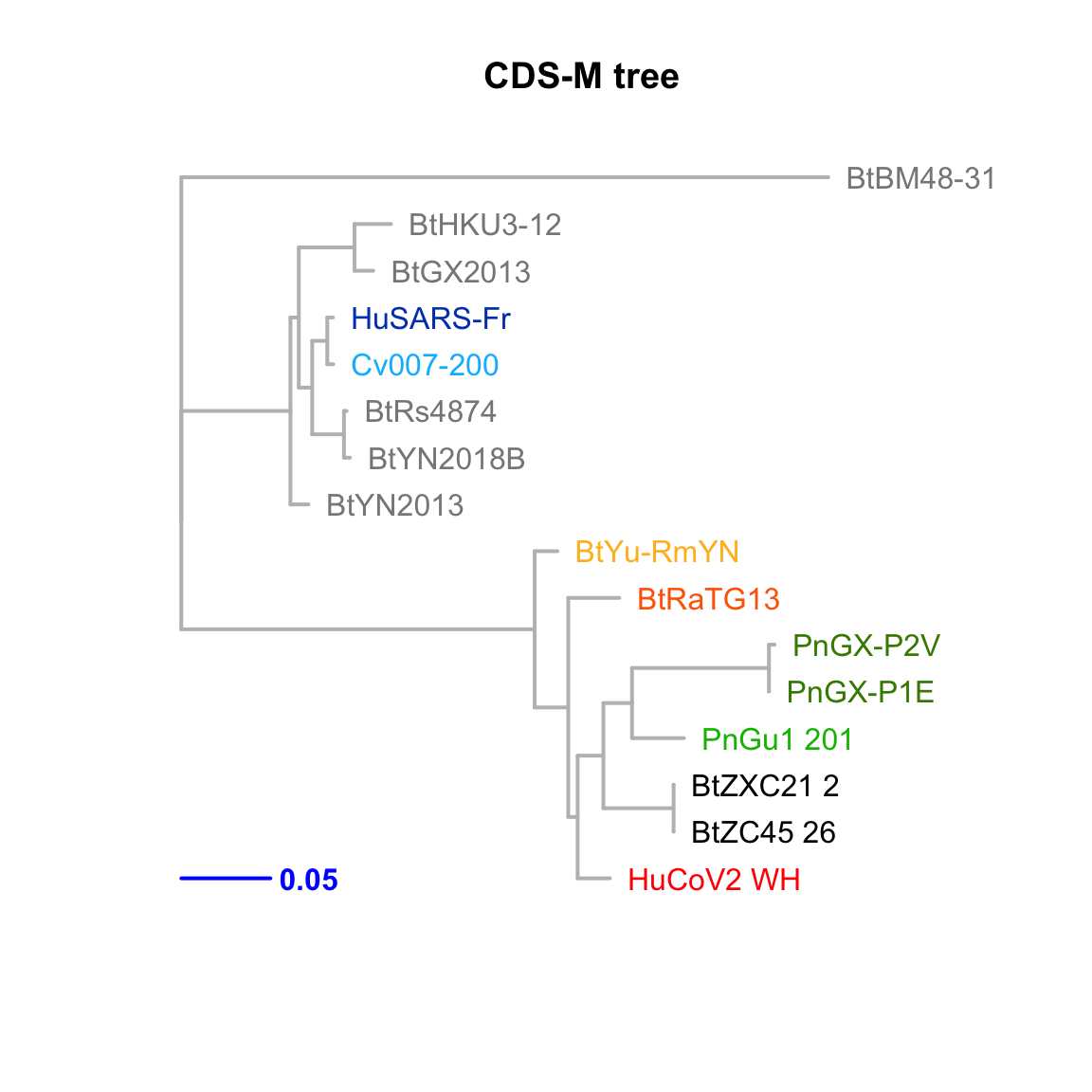
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF6
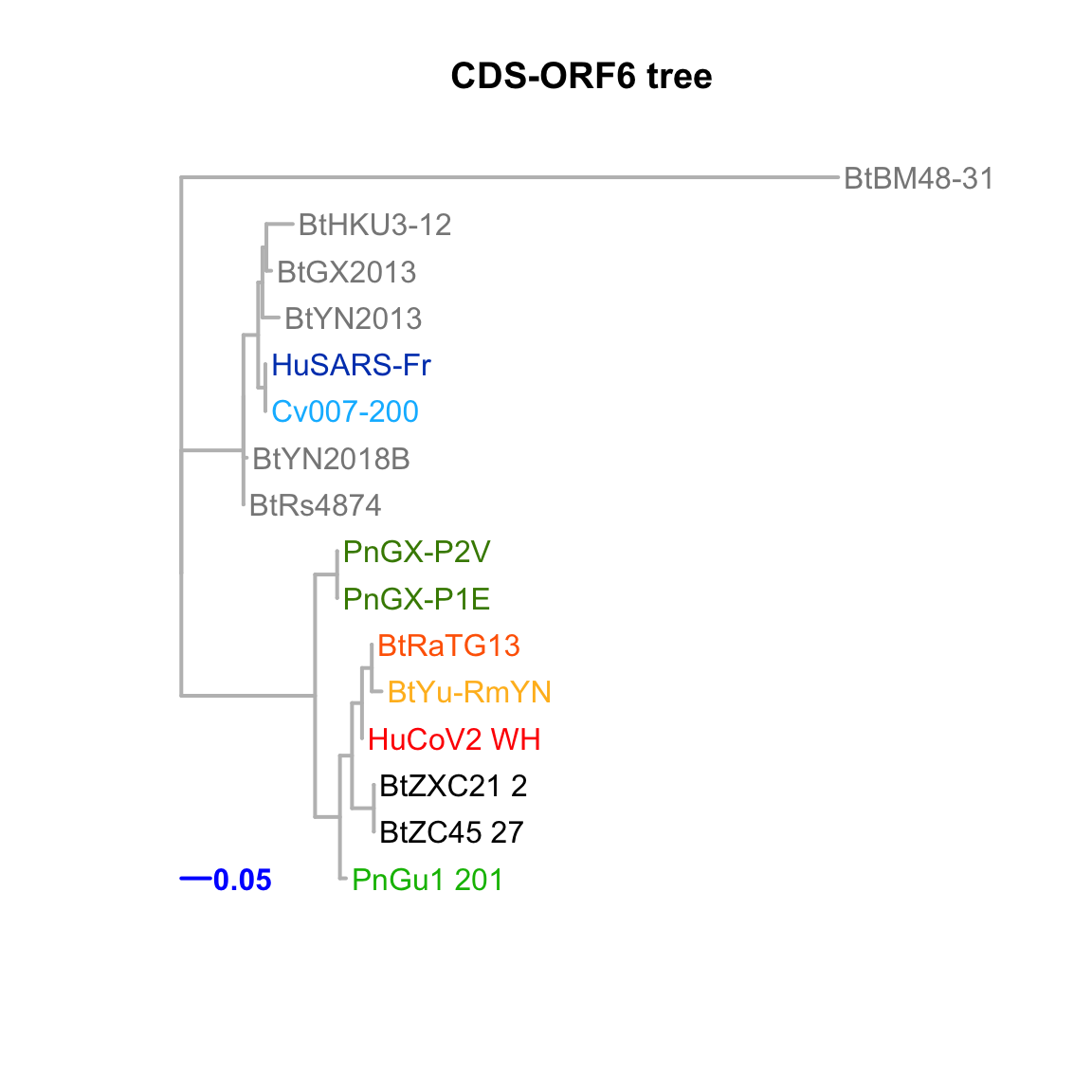
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF7a
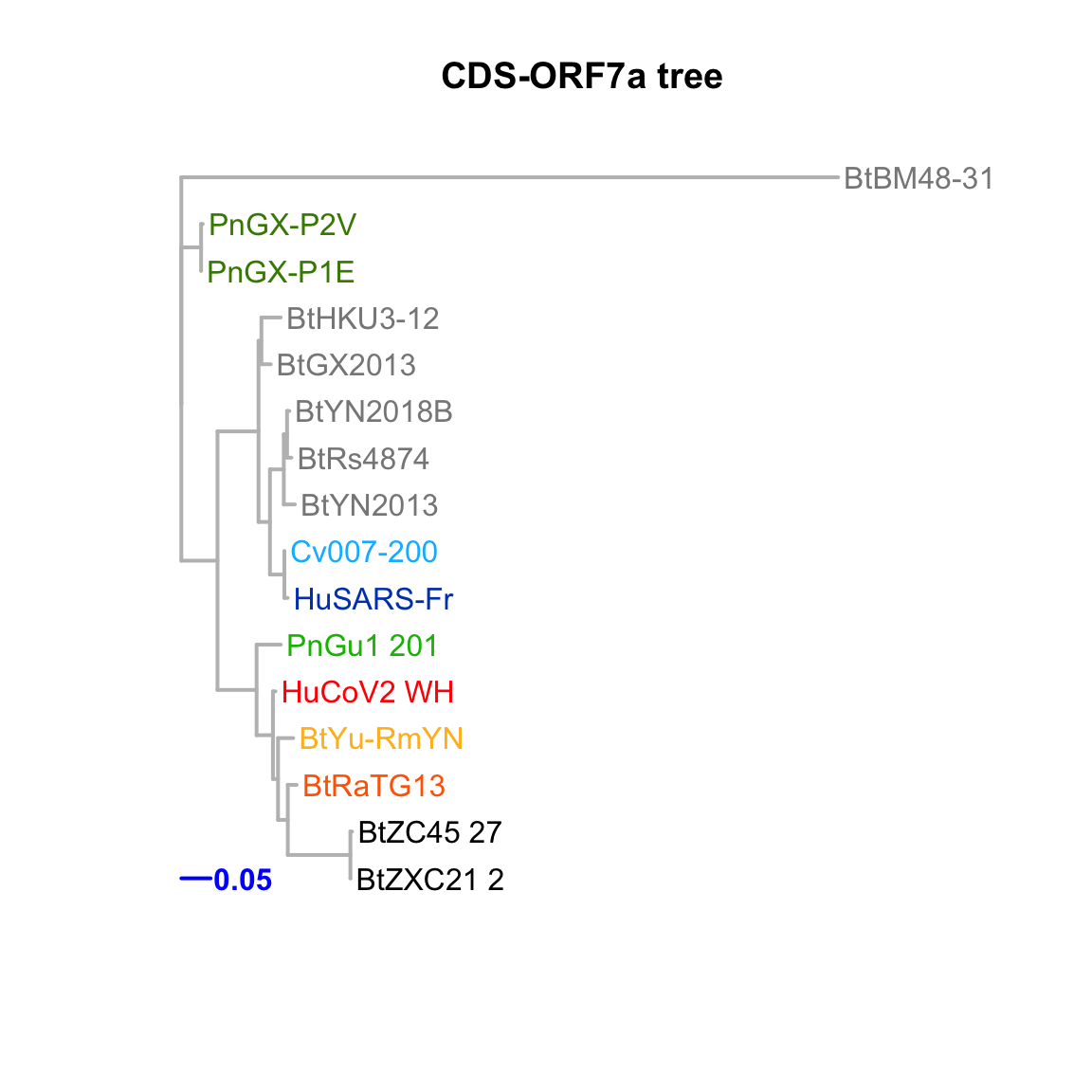
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF8
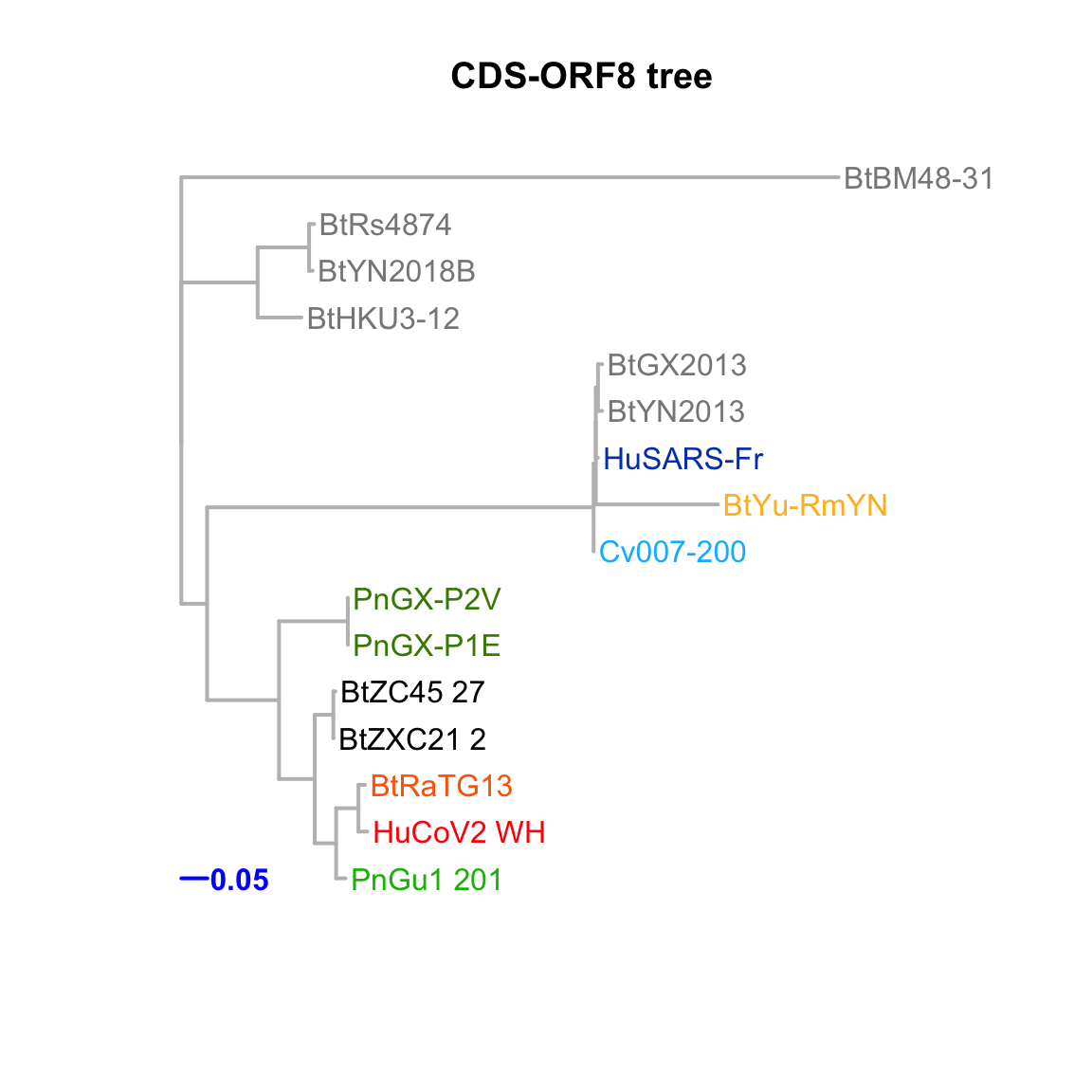
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-N
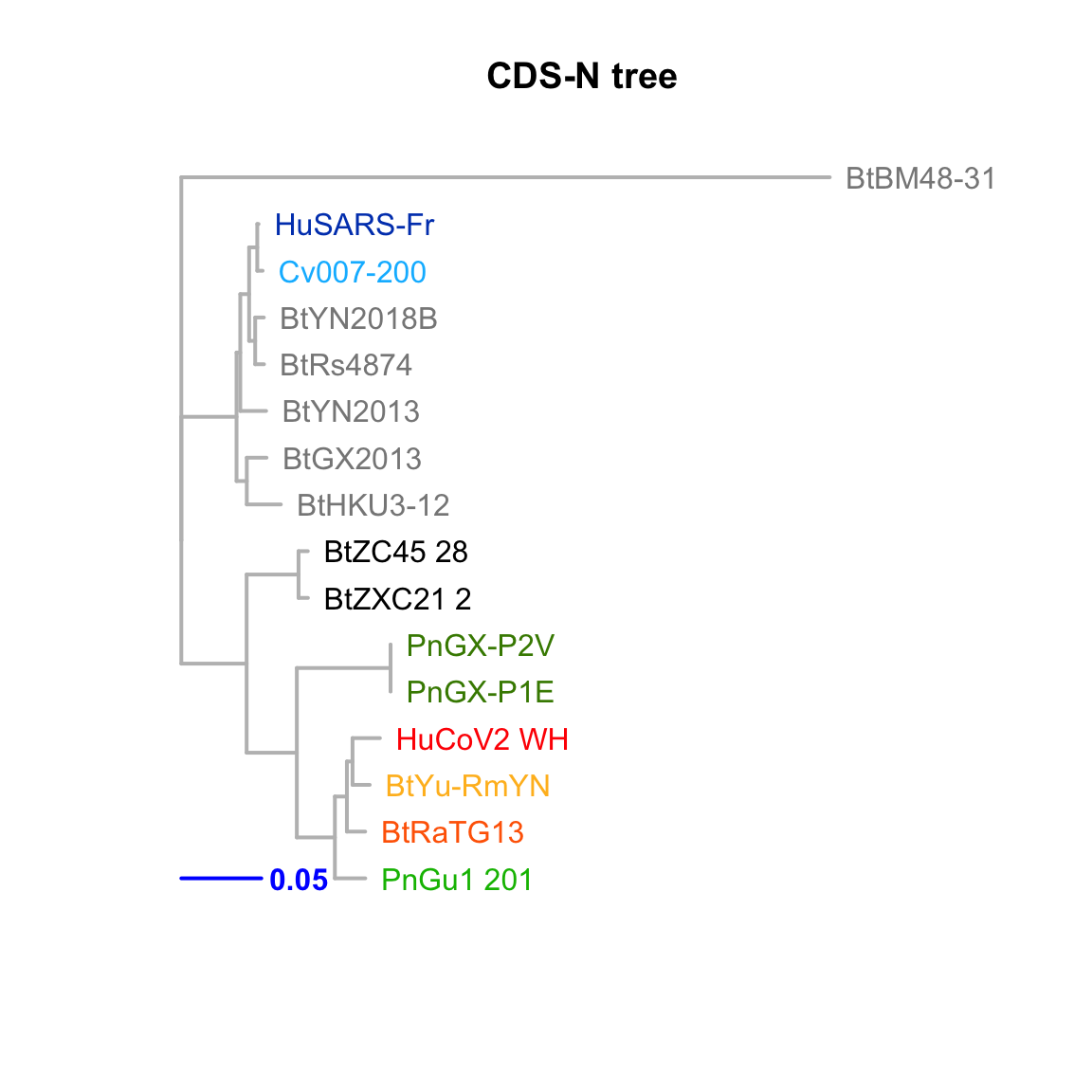
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).
CDS-ORF10
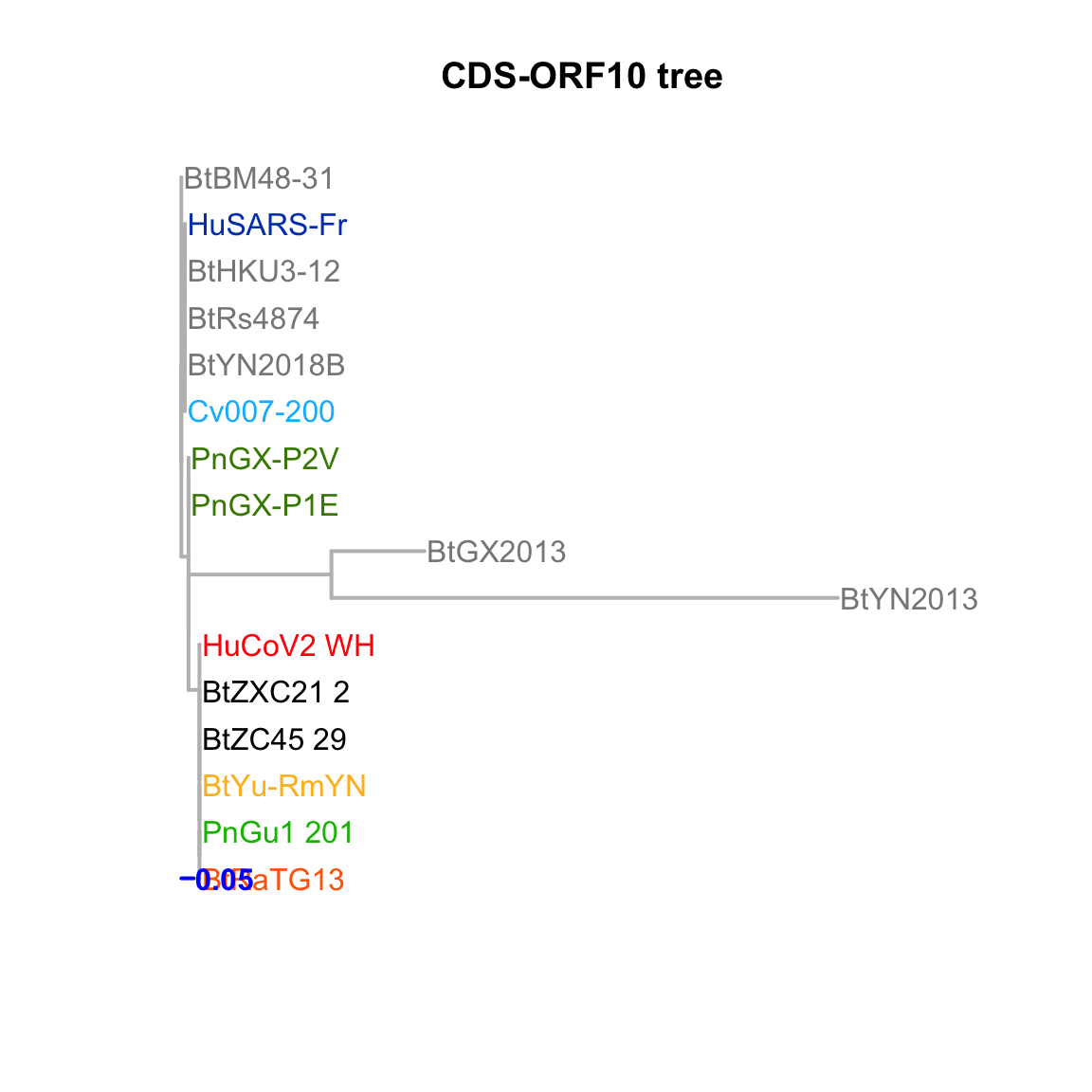
Feature-specific tree. The tree was inferred by maximum likelihood apprroach (PhyML) based on a progressive multiple alignment (clustalw).